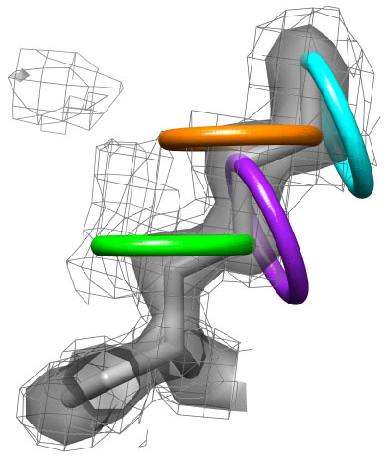
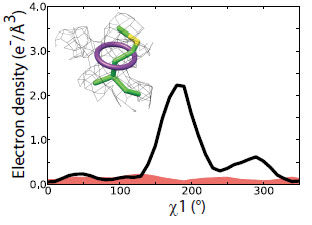
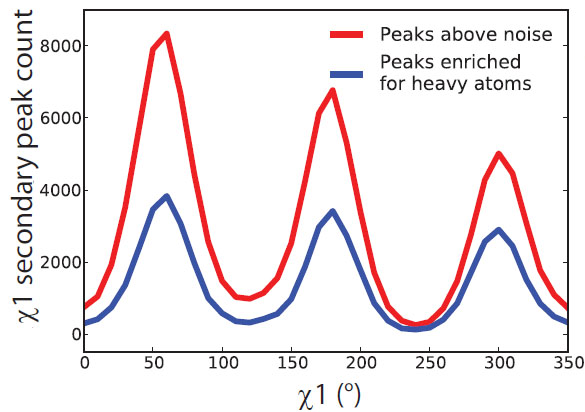
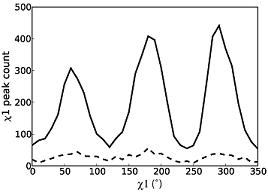
Weak electron-density features are enriched in rotameric positions. Distribution of secondary peaks (solid line) >0.3 sigma versus chi1 angle shows a trimodal distribution strongly enriched for preferred rotameric positions. The distribution of tertiary peaks (dotted line) >0.3 sigma shows a similar tri-modal distribution. |
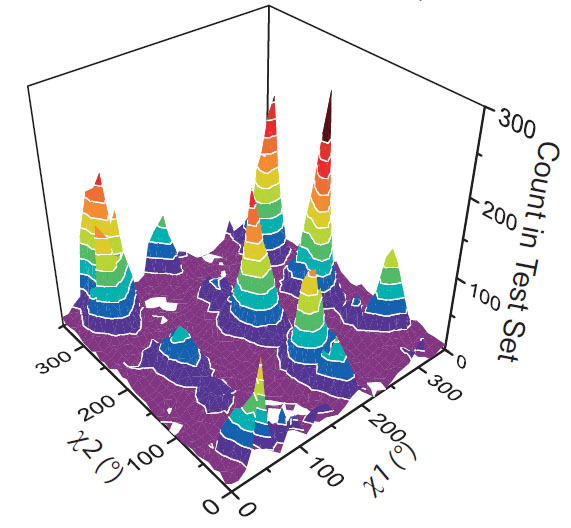
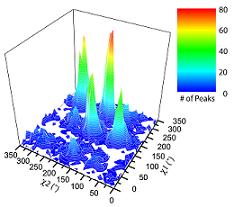
Ringer detects peaks at correlated chi1 and chi2 angles.A histogram of secondary chi1 peaks and primary chi2 peaks built from the unmodeled chi1 peaks. Crosspeaks are significantly enriched in rotameric positions (P-value < 10^-5). |
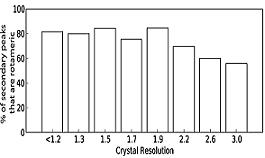
Resolution dependence of Ringer peak detection at the 0.3 sigma threshold.The percentage of secondary Ringer peaks within 30º of rotameric angles over a range of resolutions. |