nonBragg
program for calculation of absolute scattering from amorphous materials, like air, water, and protein
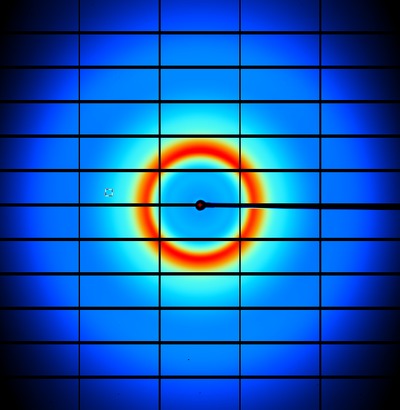


This short program calculates the absolute-scale scattered intensity from non-crystalline materials
given:
- the beam intensity in photons/s
- sample thickness in mm
- sample density in g/cm3
- molecular weight of arbitrary "molecule" in g/mol
- text file listing the structure factor of this "molecule" in electron-equivalents vs sin(θ)/λ
The definition of a structure factor
(Hartree 1925)
is the ratio of the amplitude of the wave scattered from the object of interest
(one "molecule" in this case) to the amplitude scattered by a single electron at the origin.
For a monoatomic gas, such as helium, this is simply the atomic structure factor you will find in the
International Tables of Crystallography
where it is tabulated with the same units used by nonBragg.
A few example input files are:
- He.stol- one He molecule (atom): 4 Da, 2 electrons total
- air.stol - one N2 molecule: 28 Da, 14 electrons total
- water.stol - liquid water: 18 Da/molecule
note 2.57 electrons in the forward direction
due to the long-range compressibility of water
(Clark et al. 2010)
- ice.stol - water ice: 18 Da per molecule
- nanoice.stol - nanocrystalline cubic ice, a common occurence in protein crystals
- Paratone-N.stol - a common cryo-protective oil, assuming 12 Da/molecule
The nonBragg program can also accept a diffraction image in SMV format (the native format of ADSC and Rigaku detectors),
and generate a structure factor "stol file", averaged over the image. Other X-ray detector image formats can be converted
to SMV using adxv with the command:
adxv -sa whatever_0_00001.cbf whatever_0_001.img
Feeding the stol-file written out by nonBragg back into nonBragg will
generate a perfectly centro-symmetric diffraction pattern (after correcting for polarization and detector geometry), which
can be useful for background subtraction or other background simulation tasks.
Note that nonBragg does simulate polarization and solid-angle effects from detector orientation and tilt,
but the structure factor of the material is presumed to be perfectly
centro-symmetric. This is appropriate for gasses, liquids, and genuinely amorphous solids.
If you want to simulate anisotropic structure factors that arise from oriented molecules, use
the nanoBragg SAXS example as a guide.
Don't forget to set the number of unit cells equal to one.
source code: nonBragg.c (85k) or binaries for
linux,
OSX,
Cygwin,
Windows
auxillary programs:
noisify
is a program that takes the "photons/pixel" noiseless intensity values output by nonBragg or
nanoBragg, or nearBragg as "floagimage.bin"
and adds different kinds of noise to it to generate an SMV file. This is usually faster than re-running
nonBragg just to change things like beam intensity. In addition to photon shot noise, noisify
has a few kinds of noise that nonBragg
doesn't implement, such as pixel read-out noise, beam flicker, and calibration error.
float_add may be used to
add or scale the raw "float" binary files output by nonBragg, nanoBragg, or even nearBragg
so that renderings may be divided up on separate CPUs and then
combined together. The resulting raw files may then be converted to SMV images with noisify.
float_func can perform a large number of operations on these "floagimage.bin" files.
example usage:
get nonBragg:
wget http://bl831.als.lbl.gov/~jamesh/nonBragg/nonBragg.c
compile it
gcc -O -O -o nonBragg nonBragg.c -lm -static
get some structure factor data:
wget http://bl831.als.lbl.gov/~jamesh/nonBragg/water.stol
run the simulation for a 100 micron thick slab of water and some typical beamline parameters
./nonBragg -stol water.stol -lambda 1 -MW 18 -density 1.0 -thickness 0.1 -flux 1e12 -exposure 1
view the result
adxv noiseimage.img
convert and re-scale as regular graphics file
convert -depth 16 -type Grayscale -colorspace GRAY -endian LSB -size 1024x1024+512 \
-negate -normalize \
GRAY:noiseimage.img noiseimage.png

You can confirm that the number of photons on each pixel is very similar to what you would get in reality. In fact,
using water scattering like this is one way to calibrate the flux of a given beamline.
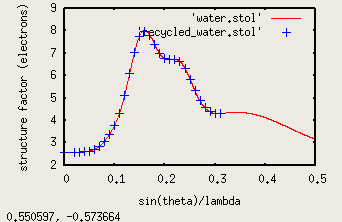
Now assume that noiseimage.img is a real diffraction pattern, you can create a "stol" file for water like this:
./nonBragg -img noiseimage_water.img -stolout recycled_water.stol -stol He.stol -lambda 1 -MW 18 -density 1.0 -thickness 0.1 -flux 1e12 -exposure 1 -nonoise
It is very important to have the right x-ray beam parameters for this to work, especially on an absolute scale!
Note that you still need to provide a "dummy" stol file to give nonBragg an idea fo the spacing you want.
The He.stol file is much coarser than water.stol, which you can see if you compare the new water stol file
to the original in gnuplot:
gnuplot
plot 'water.stol' with l, 'recycled_water.stol' lt 3
Note that the recycled_water.stol file has a much coarser sampling in this case, but if fed back into nonBragg it will
apply a cubic spline interpolation between all the points.
A fast way to compute the centro-symmetric scattering that results from averaging
over millions of protein molecules in a SAXS experiment is to compute structure factors
for a single molecule, isolated in a unit cell at least 3-4x bigger than the molecule
in all directions using phenix.fmodel, or ano_sfall.com. You can then pre-average
the structure factors in the resulting MTZ file into a "stol" file ready for input
into nonBragg with this script:
mtz_to_stol.com bigcell.pdb.mtz
which will create a file called mtz.stol that you can feed to
nonBragg:
./nonBragg -stol mtz.stol -lambda 1 -dispersion 0 \
-distance 1000 -detsize 100 -pixel 0.1 \
-hdiv 0 -vdiv 0 \
-flux 1e13 -thick 1.2 -MW 14000 -density 0.01
adxv noiseimage.img

Which starts to look more like a SAXS pattern from a conventional SAXS beamline. Note that
the "density" of the sample in this case is 0.01 g/cm^3 or 10 mg/mL.
Command-line options:
- -stol filename.stol
-
- the structure factor text list.
-
- -distance
-
- distance from sample to detector center (mm)
-
- -detsize
-
- detector size in x and y (mm)
-
- -detsize_x
-
- detector size in x direction (mm)
-
- -detsize_y
-
- detector size in y direction (mm)
-
- -pixel
-
- detector pixel size (mm)
-
- -detpixels
-
- detector size in x and y (pixels)
-
- -detpixels_x
-
- detector size in x direction (pixels)
-
- -detpixels_y
-
- detector size in y direction (pixels)
-
- -Xbeam
-
- direct beam position in x direction (mm) Default: center
-
- -Ybeam
-
- direct beam position in y direction (mm) Default: center
-
- -curved_det
-
- all detector pixels same distance from sample (origin)
-
- -oversample
-
- number of sub-pixels per pixel. Default: 1
-
- -roi xmin xmax ymin ymax
-
- only render pixels within a set range. Default: all detector
-
- -lambda
-
- incident x-ray wavelength (Angstrom). Default: 1
-
- -hdivrange
-
- horizontal angular spread of source points (mrad). Default: 0
-
- -vdivrange
-
- vertical angular spread of source points (mrad). Default: 0
-
- -hdivstep
-
- number of source points in the horizontal. Default: 1
-
- -vdivstep
-
- number of source points in the vertical. Default: 1
-
- -dispersion
-
- spectral dispersion: delta-lambda/lambda (percent). Default: 0
-
- -dispsteps
-
- number of wavelengths in above range. Default: 1
-
- -floatfile
-
- name of binary pixel intensity output file (4-byte floats)
-
- -intfile
-
- name of smv-formatted output file.
-
- -scale
-
- scale factor for intfile. Default: fill dynamic range
-
- -printout
-
- print pixel values out to the screen
-
- -noprogress
-
- turn off the progress meter
-

