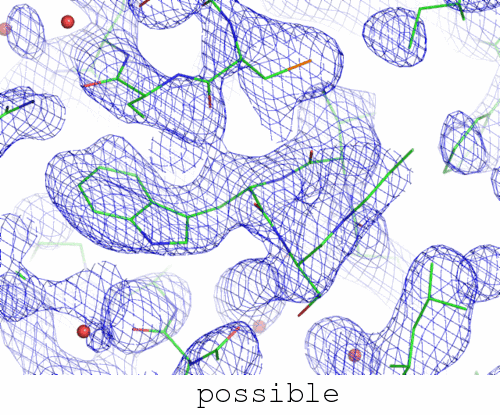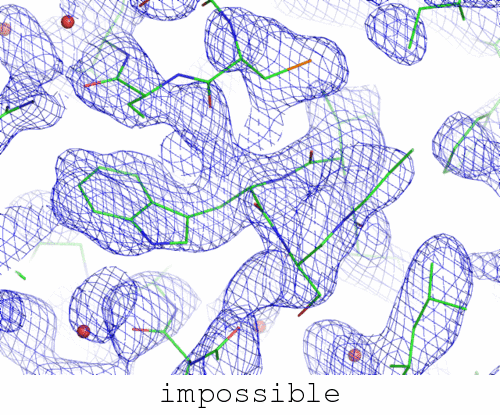
image not switching between "trapped" and "correct"? click here.
Underlying data extend to 1.0 A resolution, blue map is 2mFo-DFc contoured at 1.0x rmsd, red and green are mFo-DFc contoured at 3.5 x rmsd.
image not switching between "trapped" and "correct"? click here.
Underlying data extend to 1.0 A resolution, blue map is 2mFo-DFc contoured at 1.0x rmsd, red and green are mFo-DFc contoured at 3.5 x rmsd.
Structural biology would be far more powerful if we can get our models out of local minima, like this chain-crossing trap. If you can build a model that passes Level 10, you win!
Biological molecules adopt multiple conformations. That is how they function. And yet, ensemble refinement still has a hard time competing with conventional single-conformer-with-a-few-split-side-chain models when it comes to revealing correlated motions, or even just simultaneously satisfying density data and chemical restraints. That is, ensembles still suffer from the battle between R factors and geometry restraints. This is because the ensemble member chains cannot pass through each other, and get tangled. The tangling comes from the density, not the chemistry. Refinement in refmac, shelxl, phenix, simulated annealing, qFit, and even coot cannot untangle them.
The good news is: knowledge of chemistry, combined with R factors, appears to be a powerful indicator of how near a model is to being untangled. What is really exciting is that the genuine, underlying ensemble cannot be tangled. The true ensemble defines the density; it is not being fit to it. The more untangled a model gets the closer it comes to the true ensemble, with deviations from reasonable chemistry becoming easier and easier to detect. In the end, when all alternative hypotheses have been eliminated, the model must match the truth.
To demonstrate, I have created a series of examples that are progressively more difficult to solve, but the ground truth model and density is the same in all cases. Build the right model, and it will not only explain the data to within experimental error, and have the best possible validation stats, but it will reveal the true, underlying cooperative motion of the protein as well.
Unless, of course, you can prove me wrong?
All relevant files are available below, as a tarball, and on github.
Why can't we do this with real data? Because our models need to be untangled.
Level 1: One thing wrong: Ala39 - bond stretch
Now refine otw39.pdb against the same data as before: refme.mtz. This model has exactly the same atoms as best.pdb with exactly the same connectivity, but refined from a different starting position. The stats are notably worse: Rfree=3.4%, wE=63.5. Several geometry outliers are striking, such as the N-Cα bond of Ala39, which is 11 sigmas too long. It may surprise many that refinement does not fix this: not refmac5, not coot, not shelxl, not phenix.refine, simulated annealing or any standard protocols therein can turn otw39.pdb into best.pdb, despite them being rms 0.02 A different. otw39.pdb is trapped in a local minimum. That is the animated picture at the top of this page.
Good news is: both the difference map and the geometry outliers are screaming about where the problem is, and you only need to do one thing to fix it: the weight snap trick. After that, the model refines to a structure essentially identical to best.pdb, and has wE=18.2 again.
Level 2: One thing wrong: Val1 - clash
The model otw1.pdb is also trapped in a local minimum. Again, it has the same atoms as best.pdb, and, again, no refinement program can escape the trap. The higher stats: Rfree=3.4%, wE=24.8 arise mainly from a Molprobity clash between Val1 and a nearby water (S56). The ground truth has no clashes.

Unlike otw39.pdb, however, the weight snap trick does not work. After a weight snap, this model ends up back to where it started. What is needed here is a conformer swap trick. It is tempting to swap water S56, but that leads to two clashes, and the score rises to wE=27. The thing to do here is swap the conformers of the entire Val1 side chain and re-refine. Then there are no clashes, and the angle and torsion stats within Val1, although not bad, get markedly better after the swap. The result, as above, is identical to best.pdb.
It is because of models like this that I extended the overall score to include a term containing Rfree. But practically, as long as you don't have glaring difference peaks in your maps, wE is the most useful guide.
And, yes, I tried taking longrangetraps.pdb and cranking up the geometry weight. It didn't work. No matter the setting, longrangetraps.pdb still cannot beat best.pdb.
Level 6: Start with qFit
One of the few programs written especially for multi-conformer model building is qFit. The best model I have obtained with it is here: qfit_best.pdb, with Rfree=9.3%, and wE=97.3. Clearly some work remains to be done, and qFit is only intended to be a starting point. So, you may want to start here.
Level 7: Start with phenix.autobuild model
The output of phenix.autobuild phenix_autobuild.pdb has an excellent wE=18.1, which sounds like a champoin until you look at Rwork=17.9% and Rfree=18.4%. These are decent R factors for a normal structure, but very large when compared to best.pdb. The difference map also has huge peaks. This is because phenix.autobuild only builds one conformer. If you start here you will need to build the other one.Level 8: Start with traditional ensemble refinement
Ensemble refinement is a natural choice when you are looking for an ensemble, so I ran phenix.ensemble_refine with the deposited 1aho structure and otherwise default parameters. The output is here: phenix_ensemble.pdb. Its stats: Rfree=9.4% and wE=109.7 are similar but slightly worse than those from qFit. Using this as a starting point, however, may be tricky because the file contains 200 models.
Level 9: Long-range traps
Starting with longrangetraps.pdb, a model with wE=22.8, Rfree=3.4% may sound like a good choice, but it is deceptive. Swapping conformers of any single atom in this model does not improve wE because large stretches of the model are swapped in A-B conformation assignements relative to best.pdb. That is, if we call the correlated movements in best.pdb "jumping jacks", then longrangetraps.pdb is the "windshield wiper" model of the same average density. You will have to find a way to choose large groups of atoms without getting too many of them wrong. This model was created using simulated annealing and "back crossing", where any atoms knocked out of density or into bad geometry by the SA run were simply restored to their original positons, and the SA repeated. The result has a very good wE score, but still not as good as best.pdb.
Level 10: Let the Force Field be your guide
This is the real challenge: build a model into refme.mtz that has as-good or better R factors and validation statistics as best.pdb using your favorite model building and refinement method. The only catch is that you can't cheat. That means explaining to me (and everybody else) how to reproduce your results.
Level 11: Alternative hypothesis
This one may be impossible: Build a model into refme.mtz that is not equivalent to best.pdb but has as-good or better R factors and validation statistics. That is, find a model that conveys different correlated motions, like "windshield wipers" are to "jumping jacks", but scores just as well. Cheat if you can.
Summary table of stats:
| score | wE | Rwork | Rfree | model |
| 18.6 | 18.27 | 2.79 | 3.14 | best (cheating) |
| 21.9 | 21.36 | 3.12 | 3.48 | longrangetraps |
| 25.3 | 24.85 | 3.00 | 3.33 | onethingwrong1 |
| 59.81 | 54.58 | 5.87 | 6.57 | fused |
| 63.47 | 63.46 | 1.60 | 1.79 | onethingwrong39 |
| 85.53 | 18.13 | 17.91 | 18.42 | phenix_autobuild |
| 93.38 | 92.26 | 3.70 | 4.12 | manual_built |
| 105.1 | 103.7 | 3.94 | 4.38 | lotswrong |
| 110.5 | 97.33 | 8.84 | 9.27 | qfit_best |
| 123.5 | 109.6 | 8.87 | 9.44 | ensemble_refine |
This c-shell script untangle_score.csh implements the scoring function for this challenge, which is a weighted energy (wE). The script requies the Phenix suite, gnuplot and /bin/tcsh to be installed in order to work. Once those are set up, simply provide your refined PDB file as an argument on the command line and the script will run a variety of validation programs. It will print out the combined, weighted energy score (wE), as well as one-line summaries of the average and worst outlier for each validation metric. It should look like this:
using R/Rfree from manual_built.pdb ramalyze rotalyze omegalyze cbetadev molprobity geometry 0.183454 927 avg_bond 9.21571 48 avg_nonbond -0.303795 29237 avg_full_nonbond 3.58846 4 avg_clash 0.691348 1280 avg_angle 0.917184 564 avg_torsion 0.136204 115 avg_chiral 0.0935015 1113 avg_plane 1.86711 119 avg_omega 0.547737 96 avg_rota 0.796894 119 avg_rama 2.48353 99 avg_CBdev 4.96 -0.031 1.360 1.329 0.014 | C_63 N_64 bond 334.594 0.818 1.282 2.100 1 | HZ2_2 HG1_57 nonbond 36.4278 4x 8.85695 0.632 | A 10 AVAL HG23| A 12 ACYS SG clash 21.3 9.69 94.51 104.20 2.1 | CB_16 SG_16 SG_36 angle 10.9 -8.24 -114.36 -122.60 2.5 | C_63 N_63 CA_63 CB_63 torsion 1.52 0.25 2.26 2.51 0.2 | CA_9 N_9 C_9 CB_9 chiral 6.76 -0.052 0.052 0 0.02 | N_4 plane 12.9324 9 0 omega= 165.42 omega 4.87154 8 A 8 BASP:0.18:-87.07:-151.71:Allowed:General 0.9982 rama 5.41378 12 A 12 BCYS:0.50:0.1:253.7::::OUTLIER:OUTLIER 0.999 rota 17.64 63 manual_built :A:cys: A: 63 : 0.210: 118.44: 0.50:A: CBdev weighted energy (wE): 92.2579 1.23 2.37 334.594 4.96 21.3 10.9 1.52 6.76 14.58 4.87154 5.41378 17.64 9.21571 0.183454 0.691348 0.917184 0.136204 0.0935015 1.86711 0.796894 0.547737 2.48353 manual_built 93.3762 92.2579 3.701 4.115 n/d n/d n/d 0 1.23 2.37 334.594 4.96 21.3 10.9 1.52 6.76 14.58 4.87154 5.41378 17.64 9.21571 0.183454 0.691348 0.917184 0.136204 0.0935015 1.86711 0.796894 0.547737 2.48353 manual_built n/d n/dThe first word on each line is the unweighted energy (squared sigma deviate) associated with the metric named as the last word on each line, so you can sort -g the output to see what needs fixing the most. The stuff in between is: deviate, model, ideal, sigma, then abbreviated info on which atoms are involved. The very last line lists all validation stats at once. The rationale here being you can run a bunch of jobs on a cluster and then use the last line of the log files to sort the results. The first word of the last line is the combined UNTANLGE Challenge score. 2nd word is wE, then Rwork, Rfree is the 4th word, Molprobity score is 9th, etc.
If you have trouble running this script, please try re-running it like this:
chmod a+x ./untangle_score.csh ./untangle_score.csh manual_built.pdb debug=1 > manual_score.log tar czvf send_to_holton.tgz tempfile* manual*And then send me that send_to_holton.tgz file, along with any information you might have about which operating system you are using. You can also send your prized model to me and I can run this for you.
There are many validation metrics for macromolecular model quality, and they all have different units. Here I posit a combined, weighted energy (wE) score to combine and balance them so that trade-offs can be judged automatically. For bond, angle, plane, torsion and chiral center deviations, I extract a statistical potential from phenix.geometry_minimization. This energy is:
![]()
where the sigma (σ) (taken from Phenix) is the rms deviation expected of the bond, angle, etc. v0 is the ideal value and v the value in the model. The ground truth here has no significant deviations from ideality. For non-bonded interactions, I use a Lennard-Jones 6-12 potential, wich has a much stronger penalty for clashes than the 4th-power anti-bumping typically used in refinement. I also add an extra penalty for each Molprobity clash. The ground truth here has no clashes. I further add an extra penalty for twisted peptide bonds (omega). The ground truth has no peptides twisted more than 9 deg. The probabilities output by molprobity.ramalyze and molprobity.rotalyze are converted into a sigma-scaled deviate and therefore energy using the inverse error function. The ground truth has three unpopular rotamers. C-beta deviations also contribute a statistical energy with a presumed sigma=0.05 A. In this way, all validation metrics are all converted into statistical energies, which are now on the same scale.
There is a problem, however, with statistical energies, and that is that they tolerate huge outliers in large systems. For example, the odds of a 6-sigma bond deviation occuring by chance are about 1 in 500,000,000. That basically never happens. And yet, if 36 1-sigma deviations can be reduced to zero by creating a 6-sigma deviate, a statistical energy considers that an even trade-off. That makes no sense to me, so I weight each energy by considering the probability that it could have occurred by random chance. That is, 6-sigma deviates get a weight of 1.0, and all 1-sigma deviates get a weight near zero. More precisely, I use a
χ2 test
for the average/sum of each energy type, and for the maximum-deviating outlier in each category I use the frequency of the largest Gaussian deviate that occurs in N trials:

Where Pnn is the "probability its not noise", erf() is the integral over a normalized Gaussian, and Nthings is the number of bonds, angles, etc in each category.
A few instructive examples of Pnn are warranted:
If there is only one bond, and it is deviating from ideality by 0.67x sigma, then that energy (0.67^2 = 0.45) gets a weight of 0.5, for a total wE contribution of 0.45*0.5 = 0.22. But, if 0.67-sigma is the worst of two bonds, that gets a weight of 0.25. If it is the worst of 5, then the weight is 3%, and if 0.67-sigma is the worst outlier of 10 bonds, that gets a weight of 0.1%. On the other hand, a 4-sigma outlier gets a weight of 1.0 unless there are quite a few other bonds. If there are 10,000 bonds, you expect to see a random biggest-deviate of 4 sigmas or more about 53% of the time. So, 0.53 becomes the weight. The result is that wE tends to be dominated by the worst outliers, but random fluctuations of not-so-bad things cannot mask them.
One problem with Pnn, however, is that for large Nthings it becomes very very sharp. Sharp enough that the uncertainty in the exact value of the sigma deviate itself can dominate a decision. To make Pnn reflect the finite precision of sigma deviates I replace Pnn in practice with a 5th-power polynomial that passes through the point where Pnn=50%. So, the sigma deviate that gets a weight of 0.5 is given by Pnn, but the overall curve is smooth.
Another problem with these energy-based weights is that very large outliers tends to wash out all other considerations. For example, a worst outlier going from 100 sigmas to 99 sigmas subtracts 199 energy "points", which is far more than the 7 "point" penalty of increasing a 3-sigma outlier to 4. This is not so bad in principle, but the problem is that real structures do contain actual outliers that are supported by the density, like the three rare rotamers in the ground truth of this challenge. Genuine outlers should not confound optimization of the 2nd-worst thing. So, to curtail this effect, all unweighted energies greater than 10 are replaced with a logarithmic dependence:
![]()
This is a smooth and monotonic truncation that allows optimization in the context of outliers. The wE score can therefore be interpeted as 10x the number of potentially worrisome things in the structure. The full equation for wE in each validation category is therefore:
![]()
and the wE reported by the script is simply the sum over all categories: bonds, angles, torsions, planes, chirality, non-bond, rotamers, ramachandran angles, omega peptide bond twists,
and CB-deviations.
Finally, for an overall Challenge score, Rfree must be considered. If you have gigantic difference features in your Fo-Fc map, then you have not arrived at the correct ensemble. Strictly speaking, Pnn and χ2 could be used on difference map features, but for now I simply add a noise-offset energy derived from Rfree to wE to form a combined UNTANGLE Challenge score:

Where σR is the Rfree expected due to experimental error only. In the implementation here σR = 2%. Although algebraically unorthodox, the shape of this function approximately matches a χ2-weighted Rfree, in practice it serves only to penalize structures that have not been fully built into density.
The aim of this challenge is not water torture, so I declare here that it is not cheating to use the prior knowledge that the protein ensemble is exactly 50:50. You may also use the correct water occupancies in ground_truth_water.pdb. Refmac does not refine occupancies by default, but Phenix does. So, to turn off occupancy refinement in Phenix, use this input file noocc.eff. Getting the solvent right is a challenge all unto itself, but for now lets focus on the protein.
swapconf.csh refmacout.pdb A56 CBwill swap the conformer lettters assigned to the Cβ atom of residue 56 in chain A and write the result to a new file called swapped.pdb.
Full-matrix refinement
The refinement program SHELXL is unique in its ability to perform so-called "full matrix" refinement, which is a much more robust yet far more computationally intensive minimization method. I tried this on best.pdb and lotswrong.pdb, but did not see more than marginal improvement. For those interested in pursuing this route, the input files for shelxl refining best.pdb are here and lotswrong.pdb are here.
Worse data
Changing the resoluton of the test data by various means, such as blurring, cutting, or otherwise further corrupting the data has not had any impact on escaping these traps. They are still there no matter how bad the data are. Adding more noise to make the R factors higher also doesn't help. This is why I decided to make the test data go to 1.0 A resolution. Might as well make it fun to build!
Some things that have not been tried
phenix.rebuild_predicted_model
The latest version of Phenix allows you to use Alphafold2 predictions in model building using the already-built model as a template. If you start with the most widely-separated conformers and go from there, will it be able to predict the stuff thats harder to see? I have not tried this.
The tangling phenomenon in macromolecular ensemble refinement is analogous to these red and blue ropes.

If the ropes are the same length as the distance from floor to ceiling, then straight up-and-down is the global minimum of mechanical strain (left). If, however, the ropes take a different route, they form the simplest possible type of tangle (right). Escaping this trap cannot be acomplished with minimization (pulling on the ropes). Instead, the system must be re-built without the tangle. In the case of ropes, it is van der Waals forces that prevent the strands from passing through each other. In the case of atomic models, it is not van der Waals, but the density fit energy that keeps the chains from passing through each other.
I can't believe how many years it took me to notice this, but two or more atoms in the same blob of density have a really hard time passing through each other. This is because the calculated electron density at the moment the two atoms are on top of each other is up to twice as high and twice as sharp as it is when they are separated. This poor agreement with the observed density is a significant barrier to optimization. As you can see in this figure:

No matter what you call the two atoms: A and B, or B and A, together they will fit into oblong density. But, if you build A,B and the geometry term is better as B,A then you are stuck. The density fit term of the optimization prevents them from crossing over. So, when you split a residue into two conformers, unless you get the AB vs BA thing right at that very moment, the refinement program is not going to be able to "figure it out" down the road.
Sometimes weakening the density term with the weight snap trick can allow the barrier to be surmounted, and simulated annealing might get lucky. The conformer swap trick is an effective way to tunnel through the barrier, but there are a very large number of combinations of atoms to swap. Do you have any better ideas?
The structure factors in refme.mtz were calculated form an idealized version of the structure in 1aho. This particular PDB entry was selected because it is small, making refinement and other tasks fast, high-resolution to maximize probability of success, it has at least one instance of all 20 amino acids, it has disulfide bonds, and nothing else unusual. The ground truth model here is an ensemble, but to keep this challenge as simple as possible it is an ensemble of two.
A hand-built 2-conformer model was refined loosely against the deposited 1aho data with restraints heightened to optimize validation scores. B factors were all isotropic and refined with elevated restraints. After this optimization the real data were discarded and the ground-truth structure factors in refme.mtz were calculated from the idealized ensemble model. In other words, this challenge posits that the genuine underlying ensemble is chemically reasonable, even if we don't currently have the technology to extract it.
In the ground truth of this challenge, there are exactly two copies of every atom, including the 79 waters. Protein occupancies are 50:50 and the waters are all 50% occupied or less to keep them from clashing in refmac. The bulk solvent is flat and generated using phenix.fmodel with k_sol=0.42 b_sol=45 and otherwise default parameters. So, to match this in refmac you want to use solvent vdwprobe 1.1 ionprobe 1.1 rashrink 0.9, and for SHELXL you will want to import solvent4shelx.fab using the ABIN feature.
One detail to note: the ground truth model has hydrogens, but not all of them. Water molecules are just oxygen, and the often disordered OH hydrogens of Tyr, Thr and Ser are not included. The variable HE2 of HIS side chains is also not included. It is not cheating to leave these hydrogens out of your model.
Finally, Gaussian noise was added to the structure factors using the deposited SIGF values from the real 1aho data as widths for the random number distributions. Reflections missing in the deposited data are also missing in the simulated data. The simulated data were then named F,SIGF in refme.mtz, and the uncorrupted phases included in both PHI/FOM and Hendrickson-Lattman coefficients. The noiseless structure factor amplitudes are also included under the label F0. You may use them, but it won't make much difference.
You are, however, allowed to cheat with the water. Feel free to use the ground truth positions and occupancies of ordered solvent in ground_truth_water.pdb. Solvent structure is indeed a source of error, but for this challenge we aim to isolate the error of interest: local minima due to tangled protein chains.
Why didn't I just make a "dry" ground truth? Because clashes with waters help narrow down the possible choices. So, knowing the ground truth of the water should make solving this challenge easier, not harder. Just keep those water cannons under control.
Further peeking at the ground truth is prohibited until you get to Level 11. At this level you may cheat if you can. This is because I think Level 11 is impossible. Nevertheless, it remains a widely-held belief that any blob of density can be "over-fit" if your model has enough copies. This is that old observations/parameters chestnut, which is true for polynomials, but I maintain it is not for chemically reasonable molecular models.
Don't believe me? Try it! I encourage you and everyone you know to prove me wrong.
{kind=link}