END and RAPID Users Manual
Authors:
P. Therese Lang
James Holton
James Fraser
Tom Alber
Last updated December 2013
1.1. Why use absolute-scale and noise maps?
Electron density maps are contoured on a relative scale because X-ray crystallographic diffraction experiments cannot measure a key, forward-scattered reflection that is swamped by the transmitted beam. The structure factor of this reflection, F000, is equal to the total number of electrons in the unit cell, including the contribution from disordered solvent. Because crystals differ in composition, the absence of F000 puts each map on a different scale. The standard practice to circumvent this limitation is to represent electron density in relative units of the root mean square (RMS) deviation of map values from the mean density. These “σ-scaled” maps are sufficient for structural modeling, but it is difficult to determine which density features are signal versus noise because the σ unit has little to do with the uncertainty in the electron density. It is also impossible to quantitatively compare features in different maps, because the scale and offset relating σ to the absolute electron density varies among crystals of different molecules or even of the same molecular species with different symmetries or crystallization solvents.
To overcome this classic limitation, we developed a general computational method to calculate F000 and render maps on an absolute scale of electron number density (END) in units of electrons per cubic angstrom (e-/A3). END maps were calculated by scaling the experimental structure factors (Fobs) to structure factors calculated from the model (Fcalc, which are intrinsically on an absolute scale) and adding the average electron density of the crystal (including bulk solvent and the ordered model) to each map voxel. In contrast to σ-scaled maps, where zero corresponds to the average electron density, in END maps, zero corresponds to vacuum.
With the electron density maps on an absolute scale, we searched for an approximate electron density threshold to distinguish signal from noise. The noise level at every position in the unit cell was determined by empirically propagating errors from the structure factors into the electron density map. In this general, analytical approach, which we call refinement against perturbed input data (RAPID), errors in the experimental measurements (σ(Fobs)) or the model (|Fobs-Fcalc|) were used to add simulated noise to Fobs before re-refining the structure. Over several trials using different random number seeds, the RMS change in electron density observed at each point in the map in response to the changes in Fobs was used to calculate a “RAPID map” of the spatial distribution of errors in the electron density.
We find that noise varies with position and is generally 6-8 times lower than thresholds currently used in model building. Analyzing the new electron density maps from 485 representative proteins revealed unmodeled conformations above the noise for 45% of side chains and a previously hidden, low-occupancy inhibitor of HIV capsid protein. Comparing the electron density maps in the free and nucleotide-bound structures of three human protein kinases suggested that substrate binding perturbs distinct intrinsic allosteric networks that link the active site to surfaces that recognize regulatory proteins. These results illustrate general approaches to identify and analyze alternative conformations, low-occupancy small molecules, solvent distributions, communication pathways and protein motions.
Analyzing the new electron density maps from 485 representative proteins revealed unmodeled conformations above the noise for 45% of side chains and a previously hidden, low-occupancy inhibitor of HIV capsid protein. Comparing the electron density maps in the free and nucleotide-bound structures of three human protein kinases suggested that substrate binding perturbs distinct intrinsic allosteric networks that link the active site to surfaces that recognize regulatory proteins. END and RAPID maps enable a unified, quantitative interpretation of electron density that reveals not only low-occupancy ligands, but also dynamic structural features and alternative solvent constellations. By analogy to the Beer–Lambert–Bouguer Law in spectroscopy, which defines the relationship between molecular concentration and optical absorbance, END maps report the concentration of scattering electrons at each point in space. A current challenge remains to automatically model alternative conformations . This new information about structural distributions in crystals increases the power of x-ray crystallography to facilitate inhibitor development, visualize structural ensembles and connect macromolecular motions to functions. These capabilities open windows into biologically relevant information not included in current x-ray structural models.
1) END and RAPID are dependent on the external programs CCP4 and Phenix. Go to their respective websites (http://www.ccp4.ac.uk/ and http://www.phenix-online.org/) and follow the installation instructions.
NOTE: END and RAPID were tested extensively with Phenix version 1.6.1 and 1.6.4, and less exttensively up to version 1.8.4. Other versions may behave differently, as scaling definitions changed considerably between 1.6.1 and 1.6.4.
2) Save the distribution in the directory you want it installed in. Unpack the distribution using the following command:
[user@density ~] tar -zxvf end.rapid.tar.gz
3) The directory needs to be added to your path. Add the apropriate line to either your .tcshrc, .cshrc, or .bashrc path. For example:
Using tcsh or csh
set path = (/alber/terry/end.rapid $path)
Using bash
$PATH='/alber/terry/end.rapid/':$PATH
4) The code may need to be made executable to run. If you type END_RAPID.com and you get an error message, go into the directory and type the following command:
[user@density ~] chmod +x *
Overview of END and RAPID Maps
END and RAPID maps are general methods to solve two classic crystallographic problemsputting electron density maps on the absolute scale of e-/Å3 and calculating the noise at every point in the map. These methods allow crystallographers to differentiate signal from noise and directly compare between electron density from different maps. With these maps, we can identify and analyze alternative conformations, low-occupancy small molecules, solvent distributions, communication pathways and protein motions. (1)
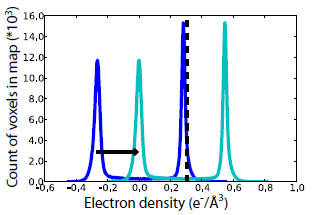
Estimation of F000 for END maps. F000 iss obtained by summing the total number of electrons in the coordinate model and the bulk solvent. An absolute scale-and-offset map for the coordinate model is obtained using the ATMMAP mode of SFALL from the CCP4 Suite (2). The mean value of this map is <ρatoms>. The structure factors of the bulk solvent mask from phenix.refine (3) were used to estimate <ρbulk>.
The histogram of the density from these structure factors (dark blue) has a mean value of zero and two peaks—one above the mean, corresponding to solvent, and one below the mean, corresponding to vacuum. The shift required to move the negative peak to 0.0 (cyan), the true vacuum level, is <ρbulk> (black dashed). To obtain the END map, “volume scale” map coefficients were specified from phenix.refine (3) and the quantity <ρatoms>+<ρbulk> was added to each voxel of the resulting 2mFo-DFc map.
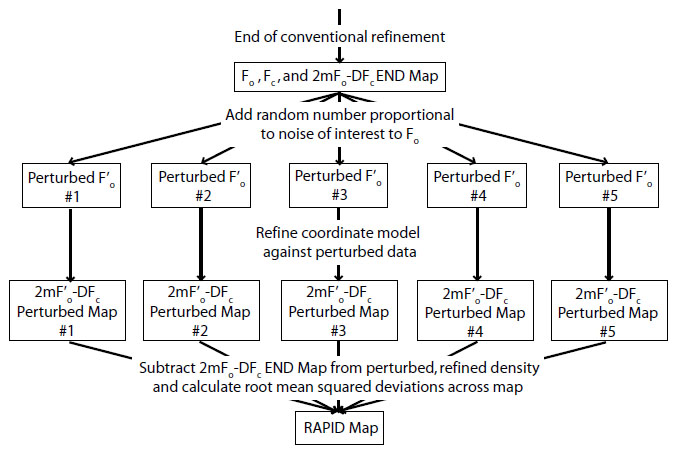
RAPID maps. The error in electron density, σ(ρ), arises from measurement errors σ(Fobs) and modeling errors (Fobs vs. Fcalc). The contribution from phase error was defined as the change in the phase from a refined model in response to a change in target amplitude (Fobs). Absolute-scale values of Fobs, σ(Fobs) and Fcalc were obtained from the phenix.refine (3) run used to generate the END map. Fobs was perturbed using SFTOOLS in the CCP4 suite (2) using the following formula,
F’obs = Fobs + r • δ
where r is a random deviate chosen from a Gaussian distribution with mean = 0 and standard deviation = 1, and δ is the expected error to be propagated into the map. Negative values of F’obs were set to zero. The new set of F’obs was used to refine the atomic coordinates in phenix.refine (3), generating a new 2mF’obs-DFcalc map (ρ’). This process was repeated five times, using different random number seeds for r. The original map (ρ) was subtracted from the five new maps (ρ’), and the RAPID map value σ(ρ) was defined as the root-mean-square (RMS) of all five ρ’-ρ values at each voxel. A flow chart of the process can be seen here:
END_RAPID.com must be run from the command line.
USAGE:
[users@density ~] END_RAPID.com phenixrefine.eff [cycles=n seeds=n cpus=n -nofofc -nosigf -norapid]OPTIONS:
phenixrefine.eff
name of the "eff" file produced by phenix.refine at your last round of refinement
cycles=n
number of macro cycles for reach phenix.refine job (default: 5)
seeds=n
do "n" parallel refinements for the RAPID maps (default: 5)
cpus=n
use up to "n" CPUs to do parallel refinements for RAPID maps (default: 1)
-nofofc
do not calculate the RAPID map based on errors from the model (|Fobs-Fcalc|)
-nosigf
do not calculate the RAPID map based on errors from experimental measurements (σ(Fobs))
-norapid
do not calculate any RAPID noise maps
END maps rescale crystallographic electron density to be on an absolute scale. RAPID maps derive from perturbing and refining the standard electron density using various noise models. The structure factors for the electron density and corresponding model must be in mtz and PDB format, respectively, and have completed at least one round of Phenix refinement (3). PDB files and structure factors for previously solved structures can be downloaded from the Protein Data Bank.
New signal revealed by END and RAPID maps currently require manual inspection and building of alternate conformers. We suggest using END and RAPID toward the end of model building and occupancy refinement to help finalize low density features. Tools like Ringer can be used to help in identifying where alternate conformations might be located. New structure added to the model as a result of END map density should be validated by small improvements in map quality and interpretability, real space correlation coefficients between Fc and Fobs, the consistency of B values and R/Rfree values.
1. Lang PT, Holton JM, Fraser JS, Alber T. Protein structural ensembles are revealed by redefining x-ray eletron density noise. PNAS USA. 111 237-247
2. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Cryst D. 2011 Apr; 67 (Pt 4):235-42.
3. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D. 2010 Feb;66(Pt 2):213-21.