What can we expect from that exploding lysozyme thing?
simulating the diffraction pattern using current XFEL capabilities
15 years ago Neutze et al. (2000)
predicted that single-molecule diffraction should be possible at X-ray Free Electron Lasers (XFELs). Well,
we've had two of them for several years now, so what's the hold up?
By using an absolute-scale simulator like nanoBragg it can be shown just how difficult this experiment is,
even with current XFEL capabilities. The prediction was that with a 100 nm wide focused beam and 1012
photons in a pulse would be enough. Now, the number of photons per pulse that LCLS can deliver depends on the
photon energy.
Lower energy gives you more photons, and also a higher scattering cross section, but if you want resolution to, say,
3 A, then Bragg's law dictates you can't go below 6 A wavelength. That is if you can catch all the back-
scattering. If you want forward-scattering and you can only get your 200 mm wide detector within 50 mm of
the sample, then you need a wavelength no greater than 3.15A to get 3A data at the edge. This is a photon
energy of 4 keV.
Based on the most recent data
it appears that at 4-5 keV you can get as much as 5x1012 photons per pulse.
As for beam size, the 100 nm focus is NOT easy to achieve, but it has been done. The fraction of photons
lost in the focusing process varies, but theoretically it can appraoch 100%, and is not worse than 50%. For
simplicity, we will assume 100% recovery here. With that, we have 5x1012 photons into a 100 nm
round area, which is 6.4x1026 photons/meter2. This is the "fluence" that nanoBragg needs.
Now that we have settled on beam and detector geometry parameters, we are ready to run nanoBragg:
compile it
gcc -O -O -o nanoBragg nanoBragg.c -lm -static
get lysozyme
getcif.com 193l
or if that doesn't work, use phenix.cif_as_mtz
refine to get the solvent parameters
phenix.refine 193l.pdb 193l.mtz | tee phenix_refine.log
Now, for single-molecule diffraction, we need to put these atoms into a very big unit cell.
It is important that this cell be
at least 3-4 times bigger than your molecule in all directions. Otherwise, you will
get neigbor-interference effects. Also, since this is a femtosecond snapshot the atoms
will not have time to move, so the B factors should all be set to very low values.
pdbset xyzin 193l.pdb xyzout bigcell.pdb << EOF
CELL 250 250 250 90 90 90
SPACEGROUP 1
BFAC 2
EOF
calculate structure factors of the molecule isolated in a huge "bath" of the
best-fit solvent.
phenix.fmodel bigcell.pdb high_resolution=2.5 \
k_sol=0.35 b_sol=46.5 mask.solvent_radius=0.5 mask.shrink_truncation_radius=0.16
note that this procedure will fill the large cell with a solvent of average
electron density 0.35 electrons/A^3. The old crystallographic contacts
will be replaced with the same solvent boundary model that fit the solvent
channels in the crystal structure.
now we need to convert these Fs into a format nanoBragg can read
mtz_to_P1hkl.com bigcell.pdb.mtz
and create a random orientation matrix
./UBtoA.awk << EOF | tee bigcell.mat
CELL 250 250 250 90 90 90
WAVE 3.14
RANDOM
EOF
and now, make the diffraction image
./nanoBragg -mat bigcell.mat -hkl P1.hkl -lambda 3.14 -dispersion 0 \
-distance 50 -detsize 200 -pixel 0.11 \
-hdiv 0 -vdiv 0 \
-fluence 6.4e26 -N 1
adxv noiseimage.img
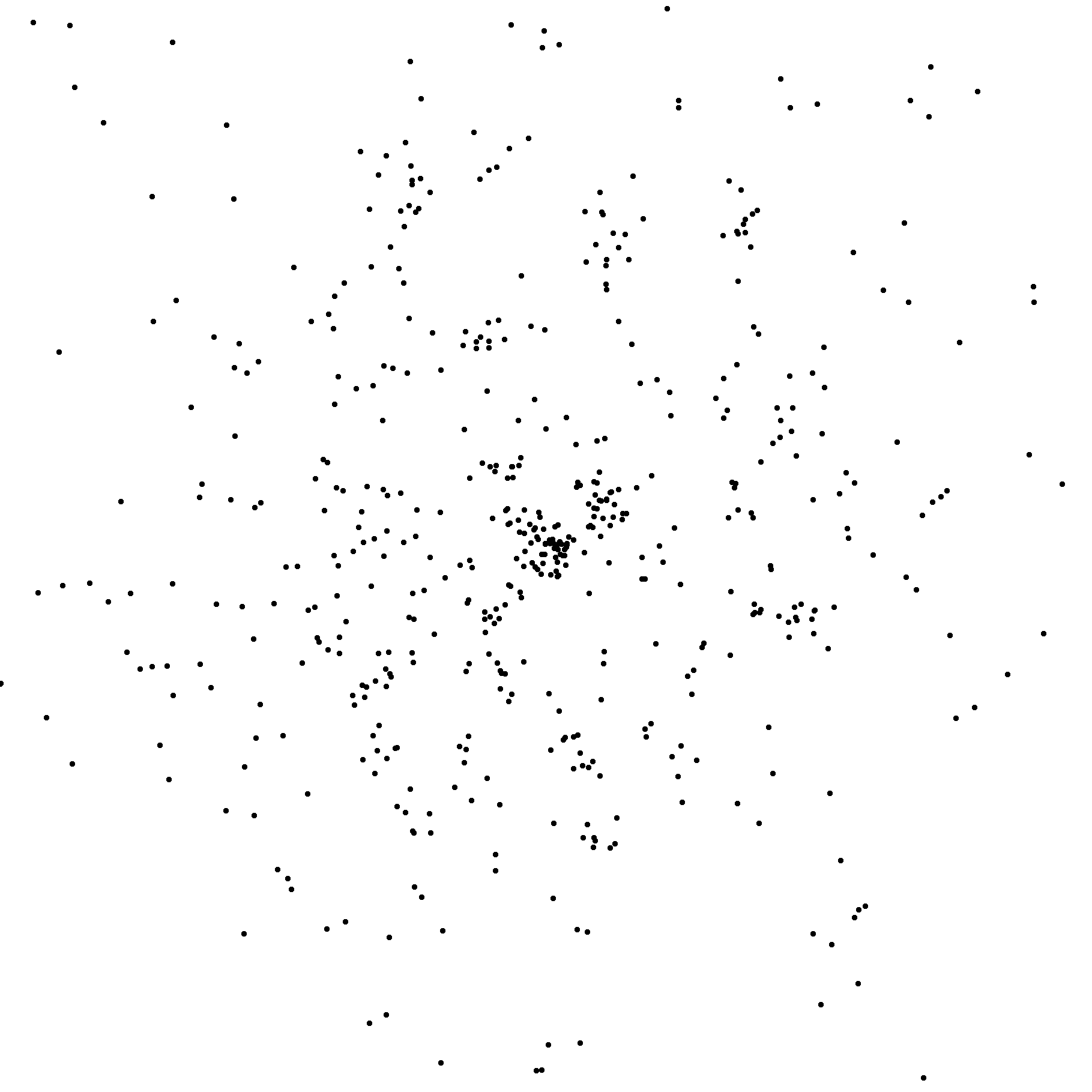
Notice that there are not a lot of photons on this image. Only about 500 in total.
In fact, I have broadened the point-spread function to make them visible.
This is the principle reason why this experiment is so hard. Software for processing
data like this has yet to be written.
What we'd like to be able to do is boost the power of the XFEL by another 3 orders of magnitude.
This is easier said than done, but if we could get 6.4x1029 photons/meter2,
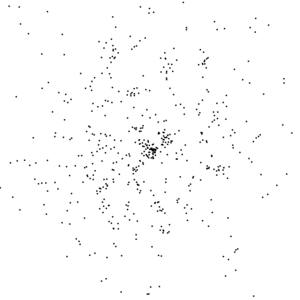
then the single-molecule diffraction pattern would look like this:
./nanoBragg -mat bigcell.mat -hkl P1.hkl -lambda 3.14 -dispersion 0 \
-distance 50 -detsize 200 -pixel 0.11 \
-hdiv 0 -vdiv 0 \
-fluence 6.4e29 -N 1
adxv noiseimage.img

There is a LOT more information in this image. Unfortunately, 3 orders of magnitude in pulse
energy is a challenge that accelerator physics has yet to find a solution to. There are limitations
on the electron density of the bunch that must somehow be overcome.
Alternatively, at least some of this 3 orders of magnitude could be accomplished by focusing. A 3
nm wide beam is 1000x smaller in area than the 100 nm beam considered here, pushing the fluence
up to the 6.4x1029 photons/meter2 needed to make the above image.
The problem,
of course, is that a single lysozyme molecule is 4 nm wide. Not only would it not quite fit in the beam,
but the "targeting problem" of hitting a 4 nm wide object with a 3 nm wide beam at a hit rate high
enough to make 3D reconstruction practical is also a challenge that has yet to be realized.
Because of all this, investigators are hoping that algorithms designed to deal with very small numbers
of photons per image can somehow recover the 3D image. This may be possible using
"manifold embedding".
But a problem with this approach is that we have lost the "single molecule" aspect of the imaging
process. It is still an averaging over billions of molecules. Just like in a crystal. And, if the
crystal doesn't diffract because the molecules are all in slightly different conformations
then the image averaged over all
these molecules taken individually will be just as blurry.
I conclude form all this that true "single molecule diffraction" will require an XFEL beam with pulse
fluence at least 1 order of magnitude higher than what can currently be achieved. And 2-3 orders of
magnitude would be nice.