image not switching between "possible" and "impossible"? click here.
image not switching between "possible" and "impossible"? click here.
I dare anyone who considers themself an expert macromolecular crystallographer to find a way to build out of this map.
Just as the legendary John Henry faced down a machine built to take his job, macromolecular crystallographers have seen a similar cyberization of their profession in the early 21st century. It seems that everything these days is automated and push-button and program developers seem to only compete on how much faster their automation package can solve a structure as opposed to someone else's automation package. Although the field is moving much faster than ever before, this situation could be considered by some to be getting depressing. Are there no genuine crystallographic challenges left for a human being to solve?
Well, of course there are. And this is one of them. The classic challenge in macromolecular crystallography is looking critically at a "ropy" electron density map and divining a helix here and a side chain there that eventually lets you build your way out to a refined structure. Yes, there are now many automation programs for doing this for you, but the important question is: are they better at it than you?
Here's your chance to prove they aren't:
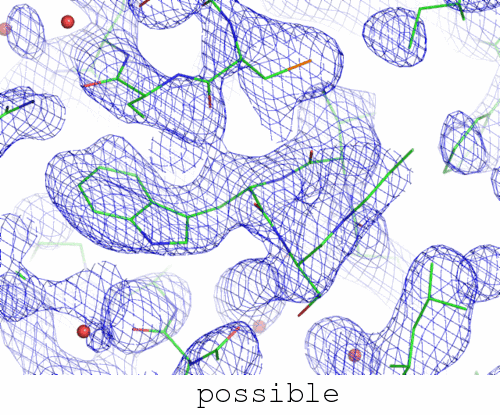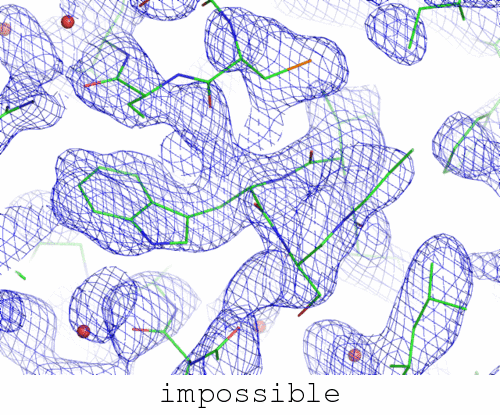
The phases in this file:
when fed with the right set of parameters into the best model building package I have available to me actually does converge to the correct structure. Specifically, the "right answer" here is the PDB entry 3dko.
However, the phases in this file:
although only slightly different than those in possible.mtz above, lead to an abysmal failure of every model-building package I have tried.
Short of actually cheating (see below), there doesn't seem to be any automated way to arrive at a solved structure from these phases. What is interesting about this is how remarkably similar these two maps are. A "representative" view of both of them is being animated at the top of this page. In fact, the correlation coefficient between the "possible" and "impossible" maps is 0.92. And yet, one can be solved automatically, and the other can't.
The question is: can you do any better?
They are actually from simulated diffraction patterns created for an educational workshop to demonstrate to novice crystallographers what a good anomalous signal and a bad anomalous signal look like. The challenge data here were made by interpolating between the "goodsignal" and "badsignal" images. This is effectively equivalent to changing the fraction of Se incorporation in the Met sites of the structure in 3dko. This particular PDB entry was selected primarily because the one long unit cell axis can be used to demonstrate spot overlap problems.
However, in the present "John Henry challenge" case, the only variable that changed between the two data sets was the fraction of Se incorporation. The difference between "possible" and "impossible" is just a 1% increment in Se occupancy. The "possible" dataset has 32% Se incorporation into the 12 methionine residues you will find in the sequence, but the "impossible" dataset only has 31% Se incorporation.
Using the right sequence information is not cheating, since that is generally something you will know before you sit down to collect data. You might also know that your Se incorporation level is ~30%.
I also don't think it is cheating to take advantage of available technology. Even John Henry had a hammer. And finding a way to work with the steam hammer instead of against it would have been healthier. However, simply feeding the sequence into BALBES is definitely cheating! This is because BALBES will simply use 3dko as a molecular-replacement search model, and since the structure is absolutely identical to the "right answer", the subsequent molecular replacement and refinement will be trivial.
And yes, this does beg another important question: how close of a homolog can you use without it being considered cheating? Well, I'd define this as the homolog that is just beyond the reach of current automation technology. I have not done a search for which starting model fits this bill, but if you can find two very closely-related structures that can and cannot be used to solve 3dko, let me know!
If you think you have found a way to build out of this map without "cheating" in any way, let me know! I will re-name the challenge after you.
James Holton <JMHolton@lbl.gov>